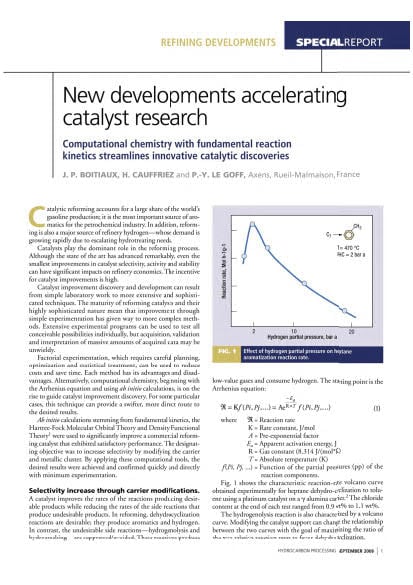
Catalytic reforming accounts for a large share of the world's gasoline production; it is the most important source of aromatics for the petrochemical industry. In addition, reforming is also a major source of refinery hydrogen whose demand is growing rapidly due to escalating hydrotreating needs.


